
In This Issue
Longevity
-
 Life's Essential 8: How a Heart-Health Score Tracks Your Biological Age
A large NHANES analysis suggests the American Heart Association's updated checklist doesn't just predict heart trouble — it lines up with how fast your body is actually aging.
Life's Essential 8: How a Heart-Health Score Tracks Your Biological Age
A large NHANES analysis suggests the American Heart Association's updated checklist doesn't just predict heart trouble — it lines up with how fast your body is actually aging.
-
 The Tobacco Aging Simulator: Seeing Your Face 15 Years Out
A new dermatologist-trained AI turns smoking risk into a portrait — predicting how cigarettes might reshape your face over the next decade and a half.
The Tobacco Aging Simulator: Seeing Your Face 15 Years Out
A new dermatologist-trained AI turns smoking risk into a portrait — predicting how cigarettes might reshape your face over the next decade and a half.
-
 Hearing the Future: Why Struggling to Follow Conversation in Noise May Be a Brain Signal
New imaging research links trouble understanding speech in noisy rooms to subtle wear in the brain's white-matter wiring — sharpening a question that matters for long-term cognitive health.
Hearing the Future: Why Struggling to Follow Conversation in Noise May Be a Brain Signal
New imaging research links trouble understanding speech in noisy rooms to subtle wear in the brain's white-matter wiring — sharpening a question that matters for long-term cognitive health.
-
 The Sirtuin–Senescence Axis: Mapping the Molecular Drivers of Aging
Three converging 2025 reviews sketch an integrated picture of why tissues deteriorate—and where senolytics, NAD+ modulators, and lysosome-restoring therapies might one day intervene.
The Sirtuin–Senescence Axis: Mapping the Molecular Drivers of Aging
Three converging 2025 reviews sketch an integrated picture of why tissues deteriorate—and where senolytics, NAD+ modulators, and lysosome-restoring therapies might one day intervene.
Performance
-
 The Algorithm That Learned to Keep Adaptive Athletes Healthy
A 40-week randomised trial of an automated, individualised prevention system points to a future where injury protocols adapt to the athlete — not the other way around.
The Algorithm That Learned to Keep Adaptive Athletes Healthy
A 40-week randomised trial of an automated, individualised prevention system points to a future where injury protocols adapt to the athlete — not the other way around.
-
 Posture, Iron, and Movement Screens: What Military Science Reveals About Injury-Proofing Yourself
Four military training studies translate into a sharper playbook for serious lifters who want to keep training — and stop leaving gains on the physio table.
Posture, Iron, and Movement Screens: What Military Science Reveals About Injury-Proofing Yourself
Four military training studies translate into a sharper playbook for serious lifters who want to keep training — and stop leaving gains on the physio table.



Peptides
-
 The New Peptide Pipeline: How AI Is Quietly Rewiring Drug Discovery
Computational design is pushing peptide therapeutics into oncology, infectious disease, and antimicrobial resistance. The early signals are real — and the caveats matter.
The New Peptide Pipeline: How AI Is Quietly Rewiring Drug Discovery
Computational design is pushing peptide therapeutics into oncology, infectious disease, and antimicrobial resistance. The early signals are real — and the caveats matter.
-
 GLP-1s Beyond Glucose: Liver, Plaque, and a Pancreas Warning Shot
Semaglutide, exendin-4, and tirzepatide are pushing GLP-1 receptor agonists into hepatology and cardiology — while a fresh case report reminds us the safety ledger is still open.
GLP-1s Beyond Glucose: Liver, Plaque, and a Pancreas Warning Shot
Semaglutide, exendin-4, and tirzepatide are pushing GLP-1 receptor agonists into hepatology and cardiology — while a fresh case report reminds us the safety ledger is still open.

Life's Essential 8: How a Heart-Health Score Tracks Your Biological Age
A large NHANES analysis suggests the American Heart Association's updated checklist doesn't just predict heart trouble — it lines up with how fast your body is actually aging.
Every few years a new longevity metric arrives with the swagger of a startup pitch — a blood panel, a saliva test, a wrist-worn gadget that promises to count down the years you have left. Most of them ask you to send a vial somewhere and trust the math. So it is a quieter kind of news when the simplest checklist on the cardiology shelf — eight ordinary habits and lab values — turns out to track, with reasonable fidelity, a credentialed measure of how fast your body is aging. That is roughly what a recent analysis of U.S. national survey data reports about the American Heart Association's updated Life's Essential 8. No vial required. Just the things your doctor already nags you about.
The study, published in Scientific Reports, pulled 17,153 adults from the National Health and Nutrition Examination Survey and asked a straightforward question: do people with higher Life's Essential 8 scores show less biological age acceleration than their birthdays would predict? After adjusting for the usual confounders, the answer was yes — higher scores were associated with a younger phenotypic age, with each step up the scale tied to a measurable slowdown in the aging clock (β = −1.22 in the adjusted model).
That is a modest effect, not a miracle, and it is an association rather than proof of cause. But it lands in a useful place for readers our age, because the eight factors are unglamorous and entirely within reach.
What the eight actually are
The American Heart Association's updated checklist covers four behaviors and four measurements: diet, physical activity, nicotine exposure, sleep, body-mass index, blood lipids, blood glucose, and blood pressure. Each is scored on a 0-to-100 scale and averaged. The average score in the NHANES sample was 68 — a polite C — which tells you something about where most American adults actually sit (Chen et al., 2025).
Two additions from the older Life's Simple 7 are worth noting. Sleep is now in the scoring, reflecting a decade of evidence that short or fragmented sleep is not a lifestyle quirk but a cardiovascular variable. And nicotine exposure now includes vaping and secondhand smoke, not just cigarettes — a sensible update for a generation whose grandchildren may be the ones bringing the vapor into the room.

Four behaviors, four measurements. None of them new — but scored together, they map onto something biology seems to notice.
The clock they used
The aging metric here is PhenoAge, an algorithm that combines nine routine lab markers — things like albumin, creatinine, glucose, C-reactive protein, and white blood cell count — to estimate a person's phenotypic age. Subtract chronological age from that and you get PhenoAge advancement: positive means your biology is running ahead of your birthday, negative means behind. In this cohort, the average chronological age was 47.5 and the average PhenoAge was 44.6, suggesting most participants were aging slightly slower than the calendar — a reminder that the metric is calibrated against a population, not a verdict (Chen et al., 2025).
PhenoAge is one of several biological-age estimators in circulation, and reasonable scientists still argue about which one best captures the underlying process. It is a credentialed proxy, not gospel. Treat the relationship as directional: higher score, younger-looking biology, on average.
No vial required. Just the things your doctor already nags you about.
What this is — and isn't
A few honest caveats before anyone reorganizes their life around a number. NHANES is a cross-section: it photographs people once, so it cannot prove that raising your score will lower your biological age. The people scoring highest on Life's Essential 8 also tend to be wealthier, better-educated, and less burdened by the kinds of chronic stress that wear bodies down independently of diet or exercise. The authors adjusted for what they could; no statistical model erases the rest.
The effect size, too, is moderate. We are talking about a measurable but not theatrical slowdown in a laboratory estimate of aging — useful as a signal, not a promise of extra decades. And PhenoAge itself is a research tool, not something your primary care doctor is likely to report at your next physical.
What the study does offer is rare and worth holding onto: a coherent link between a checklist you can actually act on and a biomarker that tries, however imperfectly, to measure the thing we all care about. That is a better deal than most of what gets sold as longevity science.

Five of the eight — blood pressure, lipids, glucose, BMI, and a candid conversation about sleep and nicotine — already live inside an ordinary annual physical.
How to read your own score
The American Heart Association publishes a free online calculator under the My Life Check banner; punch in your numbers and you get a score out of 100. For most readers in this audience the interesting exercise is not the total but the spread: a 90 in blood pressure paired with a 40 in sleep tells you where the next year's attention belongs. The strength of the LE8 framework is that it refuses to let you trade a great cholesterol number for a terrible activity number — every component carries weight.
Talk the score over with your doctor rather than your group chat. The lab values in particular — lipids, glucose, blood pressure — are where a clinician's read matters, especially if you are already on medication that flatters the numbers without changing the underlying biology.
- One checklist, eight inputs. Diet, activity, nicotine, sleep, BMI, lipids, glucose, blood pressure — scored 0 to 100.
- The link is real but moderate. In 17,153 NHANES adults, higher LE8 scores tracked with a younger PhenoAge.
- Association, not causation. A snapshot study cannot prove that raising your score will reverse biological aging.
- Sleep and nicotine now count. The updated AHA scoring reflects newer evidence on both.
- Look at the spread, not just the total. A weak component is the one worth working on next.
- Bring the numbers to a clinician. Especially the lab values — context matters more than the score.
Frequently asked questions
What are the eight factors that make up Life's Essential 8?
The American Heart Association's checklist covers four behaviors — diet, physical activity, nicotine exposure, and sleep — and four measurements: body-mass index, blood lipids, blood glucose, and blood pressure. Each factor is scored on a 0-to-100 scale, and the scores are averaged.
What is PhenoAge, and how does it differ from your actual age?
PhenoAge is an algorithm that combines nine routine lab markers — including albumin, creatinine, glucose, C-reactive protein, and white blood cell count — to estimate a person's biological age. Subtracting your chronological age from your PhenoAge gives a measure of acceleration: a positive number means your biology is running ahead of your birthday, while a negative number means it is running behind.
How strong is the connection between a higher LE8 score and a younger biological age?
The study found each step up the LE8 scale was tied to a measurable slowdown in the aging clock (β = −1.22 in the adjusted model), but the authors describe this as a moderate effect, not a dramatic one. It is also an association from a cross-sectional study, not proof that raising your score will directly reverse biological aging.
What was added to Life's Essential 8 that was not in the older Life's Simple 7?
Sleep is now included in the scoring, reflecting evidence that short or fragmented sleep is a cardiovascular variable rather than merely a lifestyle quirk. Nicotine exposure was also expanded beyond cigarettes to include vaping and secondhand smoke.
How can I find out my own Life's Essential 8 score?
The American Heart Association publishes a free online calculator under the My Life Check banner where you can enter your numbers and receive a score out of 100. The article suggests focusing on the spread across the eight components rather than just the total, since a weak component indicates where future attention would be most useful.
Sources

The Algorithm That Learned to Keep Adaptive Athletes Healthy
A 40-week randomised trial of an automated, individualised prevention system points to a future where injury protocols adapt to the athlete — not the other way around.
For decades, injury-prevention research has had a quiet credibility problem. The protocols that fill clinic walls — the eccentric hamstring sets, the proprioception drills, the load-management dogma — were largely built on data from able-bodied, often elite, often male, often very young athletes. Then they were copy-pasted onto everyone else. So when a Dutch team set out to test whether an individualised, algorithm-driven prevention system could move the needle in adaptive-sports athletes, the question was bigger than it looked. It was really a test of whether personalised prevention — the thing every sports-medicine conference has been promising for a decade — actually works when you run it through the gauntlet of a randomised controlled trial.
The trial in question, published this year in the British Journal of Sports Medicine, evaluated an intervention called TIPAS — Tailored Injury Prevention in Adapted Sports. Researchers randomised 107 athletes with physical impairments (60 to the intervention, 47 to control) and followed them for 40 weeks. Each week, athletes in the intervention arm reported their health status through a structured questionnaire. The system then fired back automated, predetermined preventive and management recommendations calibrated to that athlete's reported problems, their specific impairment, and their sport. No clinician sat in the loop on a case-by-case basis. The algorithm did the tailoring. The athlete did the work. That design alone is novel.
What the trial actually found
Here is where the performance-science geek in you needs to slow down and read the numbers carefully, because the headline and the subplot tell different stories. Across 40 weeks, the cohort logged 449 health problems — 287 injuries and 162 illnesses. Overall prevalence landed at 44% in the intervention group and 46% in controls. Pull those raw averages and you would shrug: the main effect for both injuries (OR 1.01) and illnesses (OR 1.02) crossed one cleanly, with confidence intervals that included no benefit. On a flat read, TIPAS didn't move the needle.
But injury prevention is rarely a flat read. The pre-specified analysis included a time×group interaction — the question of whether the gap between the two arms widened as the intervention had time to bite. It did. The interaction was significant at p<0.001, with injury prevalence falling in the intervention arm over time relative to control. Illness prevalence did not budge on the same axis, which is honest and worth sitting with: a behavioural, exposure-mediated outcome (injury) responded; a more biologically stochastic one (illness) did not. The rates-versus-severity analysis also flagged a significantly lower illness burden signal in the intervention arm. The full pattern is more interesting than either a win or a null.

Adaptive-sport athletes carry impairment-specific load patterns the generic prevention literature has rarely modelled.
Why the time-course matters
If you have ever periodised a training block, you already understand intuitively why the interaction effect is the more honest read. Prevention behaviours are skills. Athletes don't internalise a corrective routine or a load-modulation rule on week one — they internalise it after the system nudges them through three or four near-misses. A flat 40-week average smears the learning curve into a single number. The interaction term unsmears it. In TIPAS, the divergence between arms grew with exposure, which is exactly what a working behaviour-change intervention should look like.
This also gives a clue about why illnesses sat still. Illness in athletes is driven by sleep debt, travel, viral pressure in the training environment, and immune dips after hard blocks — variables a weekly tailored prompt can influence, but not control. Injuries, by contrast, are downstream of decisions the athlete makes inside a training session: load, technique, recovery between bouts. A nudge that arrives Monday morning and says your shoulder report is trending up, deload the press today has a much shorter behavioural latency than one that tries to prevent a winter cold.
A flat 40-week average smears the learning curve into a single number. The interaction term unsmears it.
The bigger move: prevention that scales
Step back from the effect sizes and the design itself is the story. Adaptive athletes are a population the prevention literature has historically failed: heterogeneous impairments, distinct biomechanics, sport-specific exposure patterns, and not enough of them in any single clinic to power a classical trial. A clinician-delivered, fully bespoke programme is logistically impossible to scale across that population. An automated system — one that ingests weekly self-report and returns predetermined, condition-matched guidance — is the only architecture that could plausibly reach them. TIPAS is, in effect, a proof of concept that this architecture can produce a measurable signal in a rigorous RCT.
For endurance and serious fitness athletes outside the adaptive context, the implication is not this protocol will work for you — TIPAS was built for a specific population and specific sports. The implication is structural: the next generation of prevention tools is unlikely to be a static PDF of exercises. It will be a feedback loop. You report. The system tailors. The behaviour compounds. The injury curve bends — slowly, and only if you stay in the loop long enough for the time×group term to show up in your own life.

Weekly self-report is the unglamorous engine of any tailored prevention system.
- RCT-grade evidence. 107 adaptive-sport athletes, 40 weeks, automated tailored prevention versus usual care.
- No main effect — but a real time×group interaction (p<0.001) for injuries. The benefit emerged with exposure, not on day one.
- Illnesses didn't shift on prevalence, consistent with the fact that respiratory and systemic illness is harder to nudge with weekly prompts.
- The design is the breakthrough. Automated, individualised prevention proved deliverable at scale in a hard-to-study population.
- Read the time-course, not the average. Behaviour-change interventions almost always look like late-emerging curves.
Frequently asked questions
What is TIPAS and how does it work?
TIPAS stands for Tailored Injury Prevention in Adapted Sports. Each week, athletes in the intervention arm reported their health status through a structured questionnaire, and the system returned automated, predetermined preventive and management recommendations calibrated to that athlete's reported problems, their specific impairment, and their sport. No clinician was involved on a case-by-case basis — the algorithm did the tailoring.
Did TIPAS reduce injuries overall?
The overall 40-week averages showed no significant main effect — injury odds ratios essentially crossed one with no clear benefit. However, a pre-specified time-by-group interaction analysis found that injury prevalence did fall in the intervention arm relative to the control arm over time, with that interaction significant at p<0.001, meaning the benefit emerged gradually with exposure rather than immediately.
Why did the intervention appear to affect injuries but not illnesses?
The article explains that injuries are downstream of decisions athletes make inside a training session — load, technique, and recovery — where a weekly tailored prompt has a short behavioural latency. Illnesses, by contrast, are driven by factors like sleep debt, travel, viral pressure in the training environment, and immune dips after hard blocks, which a weekly prompt can influence but not control.
How many athletes participated and for how long?
The trial randomised 107 athletes with physical impairments — 60 to the intervention group and 47 to the control group — and followed them for 40 weeks. Across that period, the cohort logged 449 health problems in total, consisting of 287 injuries and 162 illnesses.
Why is an automated prevention system considered important for adaptive athletes specifically?
According to the article, adaptive athletes have historically been failed by prevention research because of their heterogeneous impairments, distinct biomechanics, sport-specific exposure patterns, and insufficient numbers in any single clinic to power a classical trial. A fully clinician-delivered bespoke programme is logistically impossible to scale across that population, making an automated system the only architecture that could plausibly reach them.
Sources

The Tobacco Aging Simulator: Seeing Your Face 15 Years Out
A new dermatologist-trained AI turns smoking risk into a portrait — predicting how cigarettes might reshape your face over the next decade and a half.
Okay, real talk: most of us know smoking is bad for our skin. But "bad" is kind of fuzzy. It lives in the same vague drawer as "eat your vegetables" and "get more sleep." So here's the question I kept asking while reading a new paper out of the journal Dermatology and Therapy: what if you could actually see what 15 more years of cigarettes might do to your face? Not a TikTok filter. Not a carnival-mirror gag. A scientifically built portrait, trained by dermatologists, made just for you.
That's the pitch behind a new facial aging simulator described by a team of researchers and clinicians, who combined the elicited knowledge of 28 expert dermatologists with an AI image-generation model to predict how tobacco might reshape a person's face over a 15-year horizon. The work was published in 2025 in Dermatology and Therapy, and the authors frame it as a deliberate counterpoint to the viral, vibes-based aging filters floating around social media.
Here's the basic idea, explained like a smart friend who just learned it. Step one: ask a lot of dermatologists — 28 of them — to translate what they see in clinic into probabilities. Given a person's age, sun habits, BMI, sunscreen use, and how many "pack-years" of smoking they've racked up, how likely are they to hit specific stages of wrinkling, sagging, or pigmentation? Step two: feed those probabilities into an image-generation model (the team calls it AMGAN) that was trained on photos of 600 individuals scored by 15 expert graders. Step three: hand it your photo, and let it render a plausible future face.
What a "pack-year" actually means
Quick gloss, because this term does a lot of work in the paper. A pack-year is one pack a day for one year. Two packs a day for ten years? Twenty pack-years. Half a pack a day for twenty years? Ten pack-years. It's the cumulative dose, basically — the way oncologists and dermatologists measure how much tobacco a body has been swimming in over time.
The simulator uses pack-years as its main lever for tobacco. To show what the tool can do, the researchers ran a demonstration on a 43-year-old subject's photo, holding sun exposure, sunscreen, and BMI steady while varying cumulative smoking between less than 10 and more than 20 pack-years. The output: side-by-side personalized predictions of how aging signs — wrinkles, pigmentation, and the like — might progress in each scenario.

The simulator translates cumulative tobacco exposure — measured in pack-years — into a personalized 15-year facial forecast.
What if you could actually see what 15 more years of cigarettes might do to your face?
Why dermatologists, not just data
Here's the part I found genuinely clever. Pure image AI is great at noticing patterns but kind of clueless about why a face ages a certain way. A lot of internet "aging filters" basically smush a generic old-person texture onto your selfie. That's not medicine. That's a sticker.
By starting with what 28 dermatologists actually know — the probability that, say, a 50-year-old with 20 pack-years of smoking will hit a specific wrinkle grade — and then letting the image model render those probabilities onto your specific face, the researchers are trying to build something more grounded. The paper positions this approach as a scientifically validated alternative to the social-app simulators that lack transparent methodology. Translation: they're showing their work.
What this study is — and what it isn't
Okay, the honest caveats. This is a methods paper describing and demonstrating a tool, not a clinical trial proving that showing people their future face makes them quit smoking. The authors illustrate the simulator's capabilities using a single 43-year-old subject's facial image, with other variables held constant. That's a demo, not a population study.
It also doesn't tell us how accurate the 15-year predictions actually turn out to be, because — well, 15 years haven't passed yet. The probabilities come from expert consensus and a training dataset of 600 people. That's a reasonable starting point, but it's not the same as following thousands of smokers for a decade and a half and comparing the AI's forecast to their real faces.
So when you see headlines promising "AI predicts your smoking face," the more careful read is: a dermatologist-informed model can now generate personalized, scientifically grounded visualizations of plausible tobacco-driven aging. Whether those visualizations change behavior — and whether they match reality down the road — are open questions the field still has to answer.

The likely first home for tools like this: dermatology offices and public-health counseling, not consumer apps.
The bigger idea hiding in here
Step back and the interesting thing isn't really the cigarette angle. It's the template. The same framework — elicit expert probabilities, train an image model on graded photos, render a personalized forecast — could in principle be pointed at sun damage, sleep, alcohol, air pollution, you name it. Anything where the link between behavior and visible aging is well-characterized enough for clinicians to assign probabilities to outcomes.
That's a quietly big shift in how we might communicate long-horizon health risk. Numbers on a page are easy to ignore. A face that looks like yours, fifteen years from now, is harder to scroll past.
- What's new: A facial aging simulator combining 28 dermatologists' expert knowledge with an AI image generator trained on 600 graded faces.
- What it does: Predicts personalized 15-year facial aging signs based on tobacco use (measured in pack-years) plus sun exposure, sunscreen, and BMI.
- Evidence level — moderate: It's a published, methodologically transparent tool, but the demonstration uses a single subject and long-term predictive accuracy hasn't been tested against real outcomes.
- Why it matters: It turns abstract smoking risk into a concrete personalized image, which could be a powerful tool for clinical counseling and public health.
- What to watch: Independent validation, broader subject testing, and whether the same template gets applied to sun, sleep, or pollution.
- Bottom line for readers: Tools like this are educational previews, not medical advice. If you're thinking about your skin or quitting smoking, the conversation belongs with a clinician.
If you're tempted to chase down a consumer version of this — slow down. The paper describes a research tool, not an app you can download tomorrow. But the direction of travel is pretty clear: personalized, dermatologist-grounded previews of how today's habits show up on tomorrow's face. Bring that to your next derm visit, not your group chat.
Frequently asked questions
What is a pack-year, and how does the simulator use it?
A pack-year equals one pack of cigarettes smoked per day for one year, making it a measure of cumulative tobacco exposure over time. The simulator uses pack-years as its main lever for tobacco, generating personalized 15-year facial aging predictions as that number varies.
How does this simulator differ from the aging filters found on social media apps?
Unlike social-app filters that apply a generic old-person texture to a selfie, this tool starts with probability estimates from 28 expert dermatologists and renders those probabilities onto a specific person's face. The paper positions it as a scientifically validated alternative with transparent methodology, in contrast to filters that lack it.
Besides smoking, what other factors does the simulator take into account?
In addition to pack-years of tobacco use, the simulator factors in a person's age, sun exposure habits, BMI, and sunscreen use.
Does this study prove that seeing a simulated future face will help someone quit smoking?
No. The paper is a methods study describing and demonstrating the tool, not a clinical trial measuring behavior change. Whether these visualizations actually influence quitting is described as an open question the field still has to answer.
Where are tools like this expected to be used first?
The article identifies dermatology offices and public-health counseling as the likely first home for tools like this, rather than consumer apps. The authors also note that the same framework could in principle be applied to other behaviors linked to visible aging, such as sun damage, sleep, or air pollution.
Sources

Hearing the Future: Why Struggling to Follow Conversation in Noise May Be a Brain Signal
New imaging research links trouble understanding speech in noisy rooms to subtle wear in the brain's white-matter wiring — sharpening a question that matters for long-term cognitive health.
You know the moment. The restaurant is warm, the wine is good, and a friend across the table is telling a story you genuinely want to hear — except the room is loud, and somewhere between her words and your understanding, something drops. You nod. You smile. You catch about seventy percent. For years, women have been told this is simply what ears do as they age. A new line of research suggests the truer story may sit a little higher up: not in the ear, but in the brain.
Age-related hearing loss has quietly become one of the most discussed modifiable risk factors for dementia. Most of that conversation, though, has focused on the audiogram — the familiar test of whether you can detect quiet beeps. A 2025 paper in GeroScience, drawn from the long-running Rotterdam Study, argues that what happens after the ear matters at least as much. The researchers looked at central auditory functioning — your brain's ability to pull a voice out of background noise — and asked whether it tracks with the integrity of the brain's white-matter wiring.
It does. In 1,669 older adults who completed the digits-in-noise test and underwent diffusion brain imaging, poorer scores on the speech-in-noise task were significantly associated with reduced microstructural integrity in three left-hemisphere white-matter tracts: the inferior fronto-occipital fasciculus, the inferior longitudinal fasciculus, and the posterior thalamic radiation. When the authors statistically accounted for plain audibility — the beep-detection layer — two of those associations actually got stronger. The third faded. In plain language: the link between struggling in noisy rooms and changes in brain wiring is not just a side effect of muffled ears.
What the brain is actually doing in a noisy room
Hearing a friend in a crowded café is one of the most computationally demanding things your brain does in ordinary life. The ear delivers a messy acoustic stew; the brain has to separate voices, predict words, hold meaning in working memory, and ignore the clatter of plates. The tracts highlighted in the new study are part of that machinery. The inferior fronto-occipital and inferior longitudinal fasciculi are long-range cables that carry signals between regions involved in language, attention and meaning. When their microstructure degrades, the signal still arrives — it just arrives a little smudged.
That reframes a familiar complaint. "I can hear, I just can't understand" is not a contradiction. It is, increasingly, a description of central auditory function — and possibly an early read on brain health.

Understanding speech in noise draws on attention, memory and language networks — not just the ear.
"I can hear, I just can't understand" is not a contradiction. It is a description of central auditory function.
Why this matters for dementia risk
Hearing loss has been named, in major reviews of modifiable dementia risk, as one of the larger levers available in midlife and beyond. The mechanisms have always been debated: Does straining to hear deplete cognitive resources? Does social withdrawal accelerate decline? Does shared underlying brain pathology drive both? The Rotterdam Study findings nudge the conversation toward that third possibility — at least in part. The authors note that age-related declines in specific brain regions may contribute to difficulties in speech-in-noise understanding among the elderly, framing central auditory trouble as a window onto the aging brain rather than a purely peripheral problem.
It is worth being precise about what this study does and does not show. It is cross-sectional: a snapshot of brains and hearing at one moment, not proof that white-matter changes cause hearing trouble, or that hearing trouble causes dementia. It identifies an association in a large, well-characterized cohort, and it strengthens a mechanistic story. That is meaningful, but it is moderate evidence — a sharpening of the picture, not a closing of the case.
What a thoughtful reader can take from this
The practical implications are quieter than the headlines around hearing and dementia sometimes suggest. A standard audiogram in a soundproof booth may underrepresent the kind of hearing that matters most in daily life. If you have ever told an audiologist that your test came back "fine" but restaurants still defeat you, the new work offers some validation: speech-in-noise testing measures something different, and that something appears to track with brain wiring.
None of this means a difficult dinner is a diagnosis. Plenty of people who struggle in noise will never develop dementia. And the evidence that treating hearing loss prevents cognitive decline is still being built, with mixed results across trials. But the broader direction of travel — taking hearing seriously as a brain-health input, asking about speech understanding rather than just audibility, and looping in a clinician when something has changed — is well supported by where the science is moving.

Conversation is a workout for the brain — protecting the ability to have it is part of healthy aging.
- The ear is only half the story. Difficulty understanding speech in noise reflects how the brain processes sound, not just whether sound arrives.
- White-matter integrity is in the picture. In a large 2025 GeroScience study, poorer digits-in-noise scores tracked with reduced integrity in three left-hemisphere tracts.
- It isn't just muffled hearing. Two of the three associations strengthened after adjusting for audibility, suggesting a central-brain component beyond the audiogram.
- Evidence is moderate, not settled. The data are cross-sectional; they sharpen the hearing-and-brain story but do not prove cause and effect.
- Ask better questions at appointments. If restaurants are getting harder, mention speech-in-noise specifically — it's a distinct measure worth raising with a clinician.
The reassuring part of this research is that it treats women's lived experience as data. The frustration of a loud restaurant, the small social calculus of where to sit and who to face, the fatigue after an evening of straining to follow — these are not character flaws or signs of inattention. They are, increasingly, signals worth listening to. The future of hearing care, and possibly of dementia prevention, may depend on whether we start listening back.
Frequently asked questions
What is the difference between a standard audiogram and a speech-in-noise test?
A standard audiogram tests whether you can detect quiet beeps in a soundproof booth, while a speech-in-noise test measures your brain's ability to pull a voice out of background noise. The article notes that a standard audiogram may underrepresent the kind of hearing that matters most in daily life, and that the two measures capture distinct things.
What exactly did the 2025 GeroScience study find?
In 1,669 older adults who completed a digits-in-noise test and underwent diffusion brain imaging, poorer speech-in-noise scores were significantly associated with reduced microstructural integrity in three left-hemisphere white-matter tracts. When the researchers accounted for plain audibility, two of those three associations actually grew stronger, suggesting the link reflects more than just muffled hearing.
Does struggling to hear in noisy places mean I will develop dementia?
No — the article is explicit that the study is cross-sectional, meaning it is a snapshot in time and does not prove that white-matter changes cause hearing trouble or that hearing trouble causes dementia. It also notes that plenty of people who struggle in noise will never develop dementia, and that the evidence linking hearing-loss treatment to preventing cognitive decline is still being built, with mixed results across trials.
Why does understanding speech in a noisy environment feel so mentally exhausting?
The article describes hearing a friend in a crowded café as one of the most computationally demanding things the brain does in ordinary life, requiring it to separate voices, predict words, hold meaning in working memory, and ignore background clatter. The white-matter tracts identified in the study are long-range cables that carry signals between regions involved in language, attention, and meaning, and when their microstructure degrades the signal arrives a little smudged.
What should I bring up with a clinician if noisy environments have become harder to navigate?
The article suggests asking whether your hearing can be evaluated with a speech-in-noise test in addition to a standard audiogram, and whether central auditory or cognitive screenings are appropriate for your age. It recommends mentioning speech-in-noise difficulty specifically to a hearing professional or primary care clinician, framing it as a distinct measure worth raising.
Sources

Posture, Iron, and Movement Screens: What Military Science Reveals About Injury-Proofing Yourself
Four military training studies translate into a sharper playbook for serious lifters who want to keep training — and stop leaving gains on the physio table.
Every lifter I know has a ghost injury — the tweaky hip, the cranky shoulder, the ankle that rolls if you look at it sideways. We tape it, we ignore it, we PR around it. Then one day a warm-up set buries us for six weeks. Military training programs have been wrestling with the same problem at industrial scale, and a fresh batch of studies out of the French Navy, the Royal Navy, the Swedish Armed Forces, and UK veteran services offers something rare: a moderate-evidence playbook for not breaking. None of it is magic. All of it is unglamorous. And almost every piece of it maps cleanly onto how you should be training in a civilian gym.
- Your standing balance carries signal. A French Navy posturography model predicted fall-related injury with about 70% accuracy — promising, but proof-of-concept.
- A short neuromuscular warm-up beat a standard one. Royal Navy recruits saw markedly lower musculoskeletal injury rates over 10 weeks.
- Iron quietly drifts during hard training blocks. Ferritin fell in both sexes across Swedish basic training, with iron deficiency rising in women.
- Self-compassion is a recovery tool, not a soft skill. Qualitative work in UK veterans frames it as a resilience lever — small sample, big implication.
- Translate, don't transplant. Military cohorts aren't lifters; treat this as direction, not prescription.
What your balance is trying to tell you
The flashiest of the four studies put 99 male soldiers on a force plate before the French Navy Special Forces selection course, eyes closed, just standing. A neural network chewed on the sway data and tried to guess who would later wash out due to a fall-related injury. It hit about 69.9% accuracy with an AUC of 0.731 — sensitivity 56.8%, specificity 77.7%. That's a proof-of-concept, not a crystal ball, and the authors say so plainly.
For lifters, the takeaway isn't "go buy a force plate." It's that quiet, eyes-closed balance carries injury-relevant information your conscious brain doesn't. If you cannot stand on one leg, eyes closed, for 20 seconds without flailing, that's a cheap signal worth paying attention to before you keep loading the bar.

Static balance is a window onto the small stabilizers nobody trains on purpose.
The warm-up that actually moved the needle
The Royal Navy study is the one I'd staple to every gym wall. Researchers added neuromuscular pre-activation work — hip-control focused — to the warm-up of 162 recruits across a 10-week training block and compared injury rates to a 90-recruit control group running the standard warm-up. The result: musculoskeletal injury incidence dropped from 31% in controls to 8% in the intervention group, with most injuries concentrated in the lower limb and clustered in weeks 1, 2, and 5. Movement quality on the Hip and Lower-Limb Movement Screen also improved.
That's a non-randomized comparison across two cohorts, so don't read it as a guaranteed effect size — recruit populations, weather, instructors, and bad luck all vary year to year. But the direction is loud, and it lines up with everything we already believe about hip and glute activation before heavy lower-body work. Five minutes of deliberate hip-focused prep before your squat session is the cheapest insurance in the gym.
Five minutes of deliberate hip prep before your squat session is the cheapest insurance in the gym.
The iron problem nobody on the gym floor talks about
Here's the one most lifters underrate. A Swedish cohort of 58 female and 104 male recruits had bloods drawn at baseline and again after five months of basic combat training. Hemoglobin rose in both sexes, but ferritin — the storage form of iron — fell, with iron deficiency prevalence climbing significantly in women. In gender-adjusted analyses, baseline ferritin was associated with overuse injury risk, though that signal didn't survive once confounders were added. Baseline physical work capacity, meanwhile, predicted who would miss training.
Translation for the gym: extended high-volume blocks can deplete iron stores even when your hemoglobin looks fine on a standard panel. Ferritin is the better early warning, and the study suggests entering a hard block already well-conditioned matters more than people admit. None of this is a green light to start supplementing iron — excess iron is genuinely harmful, and self-dosing is a bad idea. It's a green light to ask your physician for a ferritin check if you're deep in a volume phase, training fasted often, or running low-red-meat.

Ferritin is the quiet variable in long, hard training blocks.
The recovery variable lifters refuse to train
The fourth study is the one I almost skipped, then couldn't stop thinking about. UK military veterans in recovery from alcohol use disorders were interviewed about self-compassion — the practice of meeting your own setbacks with the same patience you'd give a friend. Through an interpretative phenomenological lens, researchers identified two themes — "Searching for Safety" and "Healing with Honour" — and found self-compassion was salient in veterans' AUD recovery, though sometimes perceived as incongruent with military identity.
Five participants. Qualitative. Not about the gym. So why include it? Because the shame-spiral after a missed PR, a fumbled cut, or a tweaked back is the exact mechanism that pushes lifters into the dumbest decisions of their training career — training through pain, slashing food, doubling volume to "make up." The veterans' framing is useful: self-compassion isn't softness. It's the cognitive infrastructure that lets you make the unsexy call — deload, sleep, see a clinician — instead of doubling down. Take this one as direction, not data.
How to actually use this
None of these studies were run on civilian lifters, and the evidence here is moderate, not settled. But the convergence is hard to ignore: balance signals matter, hip-focused neuromuscular prep reduces injury, iron status quietly erodes under load, and how you talk to yourself in a setback predicts what you do next. That's a four-lever model — screen, prep, fuel, recover — that doesn't require a force plate or a flag on your shoulder to deploy.
Build the eyes-closed single-leg balance check into your warm-up. Spend five minutes on hip-activation work before lower-body days. If you're deep in a volume block, ask your physician about a ferritin panel before you assume you're just "tired." And when training goes sideways, treat the deload like the disciplined call it is, not a moral failing. Talk to a clinician about anything that hurts for more than a couple of weeks. The lifters who train for decades aren't the ones who never got hurt. They're the ones who saw it coming.
Frequently asked questions
What did the French Navy posturography study actually show, and how reliable is it?
Researchers placed 99 male soldiers on a force plate — eyes closed, just standing — before a special forces selection course, then used a neural network to predict who would later sustain a fall-related injury. The model reached about 69.9% accuracy with an AUC of 0.731, which the authors themselves describe as a proof-of-concept rather than a reliable screening tool.
How much did the neuromuscular warm-up reduce injuries in the Royal Navy study?
Recruits who used a hip-control-focused neuromuscular pre-activation warm-up had a musculoskeletal injury rate of 8% over 10 weeks, compared to 31% in the group that used the standard warm-up. Most injuries in both groups were concentrated in the lower limb and clustered in weeks 1, 2, and 5.
Why can't I just rely on a standard blood panel to know if my iron is okay during a hard training block?
The Swedish study found that hemoglobin rose in both male and female recruits over five months of basic combat training, while ferritin — the storage form of iron — fell. Because a standard CBC measures hemoglobin rather than ferritin, it can miss declining iron stores, which is why the article recommends asking a clinician specifically for a ferritin panel.
What is the one-leg balance test mentioned in the article, and what does it signal?
The article describes a simple self-check: standing on one leg with eyes closed for 20 seconds without losing control. The point is not to replicate force-plate science but to notice that quiet, eyes-closed balance carries injury-relevant information about the small stabilizers that most people never train deliberately.
Why does the article include a study about veterans and self-compassion when it's not about lifting?
The qualitative study, drawn from five UK military veterans recovering from alcohol use disorders, identified self-compassion as a resilience lever — framed through the themes 'Searching for Safety' and 'Healing with Honour.' The article applies the concept to training because the same shame-driven response to setbacks — pushing through pain, slashing food, doubling volume — leads lifters into poor decisions that prolong injuries.
Sources
- Posture analysis in predicting fall-related injuries during French Navy Special Forces selection course using machine learning: a proof-of-concept study. — BMJ military health
- Efficacy of neuromuscular exercises to promote movement quality and reduce musculoskeletal injury during initial military training in Royal Navy recruits. — BMJ military health
- Changes in haemoglobin and ferritin levels during basic combat training: relevance for attrition and injury frequency. — BMJ military health
- Courage, camaraderie and compassion: a qualitative exploration into UK military veterans' experiences of self-compassion within the context of alcohol use disorders and recovery. — BMJ military health
Bone Health Before Spine Surgery: Which Anabolic Drugs Actually Move the Needle?
A single-institution analysis used CT-derived Hounsfield Units to compare modern osteoporosis drugs in preoperative patients. The signal is real but moderate — and frailty quietly blunts the response.
Bone is the part of the quantified-self stack that almost nobody quantifies — until a surgeon says the word osteoporosis and suddenly every vertebra on the CT becomes a number. For patients heading into spine surgery, those numbers matter: poor bone quality raises the odds of screws loosening, cages subsiding, and constructs failing. A 2025 analysis in the Journal of Clinical Medicine took a pragmatic look at what actually shifts those numbers before the operating room, comparing modern anabolic and antiresorptive drugs using the most n-of-1-friendly bone metric the average patient already has lying around: the Hounsfield Unit reading on a routine CT.
The setup is the kind of real-world dataset biohackers tend to trust more than tightly controlled trials. Researchers at a single multisite institution pulled records on 267 preoperative spine surgery patients — median age 74, two-thirds female — who had been treated with one of four drugs: the anabolics teriparatide and romosozumab, or the antiresorptives denosumab and alendronate. They measured lumbar Hounsfield Units before and after treatment and asked a simple question: who crossed a clinically meaningful threshold of at least a 7-point HU improvement?
Just under half — 127 patients, or 47.6% — did. That headline number is worth sitting with. It says the modern pre-op bone toolkit moves measurable density in roughly one in two patients within a treatment window, which is genuinely useful, and also that the other half didn't clear the bar. For a Protocols-section reader, this is the moderate-evidence reality: real signal, real ceiling.
The Four Drugs, Briefly
The comparison spans two mechanistic camps. Anabolics — teriparatide (a PTH analog) and romosozumab (a sclerostin-blocking monoclonal antibody) — push osteoblasts to build new bone. Antiresorptives — denosumab (a RANKL inhibitor) and alendronate (a bisphosphonate) — slow the osteoclasts that tear bone down. In the cohort, alendronate (95 patients) and denosumab (113) were the workhorses; romosozumab (31) and teriparatide (28) were the smaller, more targeted groups, typically reserved for higher-risk bone.
The univariable comparisons showed the groups weren't apples-to-apples to begin with: they differed significantly by age, sex, BMI, frailty scores, and even baseline HU. That's the texture of real clinical practice — the sicker, frailer, thinner-boned patients tend to get routed toward the anabolics. Any honest read of the data has to account for that selection.
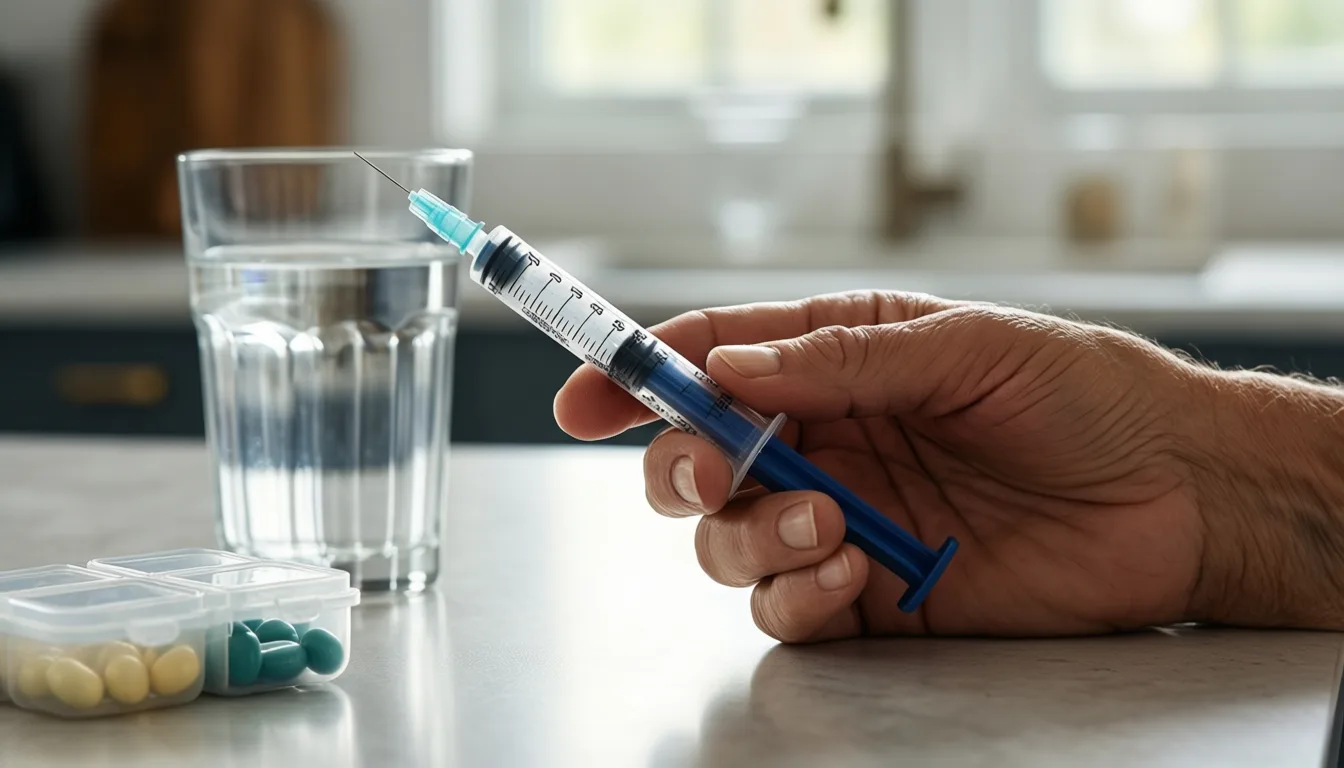
Anabolic regimens like teriparatide and romosozumab are delivered by injection, often for a fixed pre-surgical window.
Frailty Is the Quiet Variable
The most actionable finding for the quantified-self crowd isn't which drug ranked first — it's that the patient's underlying frailty appears to shape who responds. The study explicitly set out to identify factors influencing treatment response, leaning on two indices most readers won't have heard of: the modified Frailty Index (mFI) and the Risk Analysis Index (RAI). Both differed significantly across medication groups in the baseline comparisons, and both were built into the logistic regression models predicting HU gain.
The implication, framed cautiously: bone-building drugs don't operate in a vacuum. Pre-treatment HU, BMI, and composite frailty scores all carry information about who will and won't cross the 7-point threshold. For a reader optimizing before elective spine surgery, that suggests the protocol question isn't only "which drug?" but "what shape am I in when I start it?" Sarcopenia, nutritional status, and overall physiologic reserve are not side quests here — they're part of the response curve.
Bone-building drugs don't operate in a vacuum. Frailty appears to shape who actually crosses the response threshold.
What HU Actually Tells You
One reason this paper resonates with the data-obsessed: Hounsfield Units are already sitting on most pre-op CT scans, no extra DEXA appointment required. HU is a density measurement baked into CT physics; in the lumbar spine, it correlates with bone mineral density and has become a pragmatic surrogate for fracture and instrumentation risk. The investigators chose a ≥7-point improvement as their threshold for "responder," which gives readers a concrete, repeatable yardstick — the kind of pre/post number you can actually track between scans rather than a vague "bone got better."
Caveats apply. This is a single-institution retrospective cohort, not a randomized comparison. The drug groups weren't balanced. The follow-up windows and treatment durations weren't standardized in the abstract-level data. And HU change, while clinically meaningful, is a proxy for what surgeons actually care about: fewer mechanical failures after fusion. The paper is a useful map of the terrain, not a verdict.

Frailty indices factor in mobility, strength, and comorbidities — variables that respond to training, not just pharmacology.
- The pre-op bone toolkit works — about half the time. Roughly 47.6% of patients hit the ≥7-point HU improvement threshold across all four drugs.
- Anabolics and antiresorptives were both in the mix. Teriparatide and romosozumab (anabolic) sat alongside denosumab and alendronate (antiresorptive); each group served a different patient profile.
- Frailty matters as much as the molecule. mFI and RAI scores differed significantly across drug groups and were modeled as predictors of response.
- Hounsfield Units are a tractable metric. They live on routine CTs and offer a concrete pre/post number — useful for tracking, not a substitute for clinical judgment.
- This is one institution, not a randomized trial. Treat the rankings as a map of real-world practice, not a definitive head-to-head.
For the n-of-1 mindset, the most honest takeaway is that pre-surgical bone optimization is now measurable, comparable, and worth asking about — but it's also a domain where the patient's underlying biology, captured imperfectly by frailty indices, sets a ceiling on what any drug can do in a short pre-op window. Modern anabolics are powerful tools. They are not, on the evidence to hand, magic ones. Bring the CT number to the conversation, and let a clinician who knows the rest of the chart pick the protocol.
Frequently asked questions
What percentage of patients in the study actually saw a meaningful improvement in bone density before spine surgery?
Just under half — 127 patients, or 47.6% — achieved a clinically meaningful improvement of at least 7 Hounsfield Units. The study authors note this reflects a real signal with a real ceiling, meaning the pre-surgical bone toolkit works roughly one in two times within a treatment window.
What is the difference between the anabolic and antiresorptive drugs compared in the study?
Anabolic drugs — teriparatide and romosozumab — work by pushing osteoblasts to build new bone, while antiresorptive drugs — denosumab and alendronate — work by slowing the osteoclasts that break bone down. In the study cohort, alendronate and denosumab were the more commonly used options, while teriparatide and romosozumab were typically reserved for higher-risk patients.
Why does frailty matter when it comes to how well bone medications work before surgery?
The study found that patients' frailty scores — measured by the modified Frailty Index and the Risk Analysis Index — differed significantly across drug groups and were modeled as predictors of who responded to treatment. The article suggests that bone-building drugs don't operate in a vacuum, and a patient's underlying physiologic reserve, including factors like sarcopenia and nutritional status, sets a ceiling on what any drug can achieve in a short pre-operative window.
What are Hounsfield Units, and why did researchers use them to measure bone health?
Hounsfield Units are a density measurement built into CT scanning that correlates with bone mineral density and is used as a surrogate for fracture and instrumentation risk in the lumbar spine. Researchers used them because they are already present on most routine pre-operative CT scans, requiring no additional DEXA appointment, and offer a concrete before-and-after number that can be tracked between scans.
How reliable are the study's findings — was this a randomized controlled trial?
The study was a single-institution retrospective cohort analysis, not a randomized trial, and the drug groups were not balanced — sicker, frailer patients tended to be routed toward the anabolic medications. The article describes the paper as a useful map of real-world practice rather than a definitive head-to-head comparison.
Sources
- Impact of Frailty and Other Factors as Estimated by HU to Predict Response to Anabolic Bone Medications. — Journal of clinical medicine

The Glucose Swing Factor: What a New Study Says About BMI and Late-Pregnancy Blood Pressure
A multicenter analysis suggests that early-pregnancy glucose variability may be a hidden link between higher pre-pregnancy BMI and late-onset hypertensive disorders — and a possible lever for prevention.
For years, the conversation around pregnancy and blood sugar has hovered around one diagnosis: gestational diabetes. But a quieter metric — how much your glucose swings up and down across the day — is starting to get its moment. A new multicenter analysis out of Japan suggests that this kind of variability in early pregnancy may be one of the missing links between a higher pre-pregnancy BMI and late-onset hypertensive disorders, the umbrella that includes late preeclampsia. It's an early-stage finding, not a verdict. But it points to something we don't talk about enough: the metabolic terrain of the first trimester might be quietly setting the stage for what happens in the third.
- The headline finding: In 802 pregnancies across 14 centers, high glucose variability in early pregnancy partially mediated the link between higher pre-pregnancy BMI and late-onset hypertensive disorders.
- Why it matters: Late-onset preeclampsia has few good prevention strategies, so any modifiable upstream signal is worth attention.
- It's not just gestational diabetes: The association between variability and hypertensive disorders was actually stronger in pregnancies without GDM.
- What it isn't: A randomized trial, a CGM recommendation, or proof that flattening glucose curves prevents preeclampsia.
- The practical read: Pre-conception metabolic health — and a clinician-guided plan if BMI is elevated — looks more relevant than ever.
What the study actually did
The multicenter retrospective study, published in Scientific Reports, pulled data from 802 pregnancies across 14 facilities. Every participant had a 75-gram oral glucose tolerance test (OGTT) by 20 weeks of gestation — early enough to capture the metabolic baseline before the third-trimester hormonal surge that usually triggers gestational diabetes screening. Researchers then used structural equation modeling, a statistical method that maps out direct and indirect pathways at the same time, to ask a specific question: when a higher pre-pregnancy BMI is linked to late-onset hypertensive disorders of pregnancy (LoHDP), how much of that link runs through glucose variability?
The answer, in the data, was: a meaningful slice of it. Overweight and obese participants had high glucose variability at a rate of 26.1% versus 16.4% in their non-overweight counterparts, and LoHDP at 17.6% versus 7.9%. The modeling found a direct effect of BMI on LoHDP, plus an indirect effect routed through glucose variability — a small but statistically significant detour that helps explain part of the risk.

Steadier meals — protein, fiber, healthy fats — are the kind of intervention researchers have long suggested might smooth glucose curves. Whether that translates to lower preeclampsia risk specifically is still an open question.
Why glucose variability — not just average sugar
Most of us were taught to think about blood sugar as an average: A1C, fasting glucose, the OGTT pass/fail line. Variability is a different lens. It asks how jagged the curve is — the peaks after meals, the dips between them, the rollercoaster versus the gentle hill. Outside of pregnancy, glucose variability has been linked in observational research to vascular stress, oxidative damage, and endothelial dysfunction — the same biological neighborhoods where preeclampsia lives. So a hypothesis has been brewing: maybe it's not only how high glucose goes, but how restless it is, that matters for the placenta and the maternal vascular system.
This study doesn't prove that mechanism, but it adds a meaningful data point. And the most interesting wrinkle is that the variability–LoHDP association was stronger in pregnancies without gestational diabetes (β = 0.25, p < 0.001) than in the overall group. In other words, you can pass the standard GDM screen and still carry a metabolic signal that may be relevant to later blood-pressure complications. That challenges the binary framing many of us bring to pregnancy metabolism: you either have GDM or you don't.
You can pass the standard gestational diabetes screen and still carry a metabolic signal that may matter later in pregnancy.
What this doesn't mean
Here's where the magazine has to pump the brakes, because this is where the internet usually doesn't. This is a retrospective observational analysis, not a trial. Mediation modeling is powerful, but it describes statistical relationships in a snapshot of data — it doesn't prove that lowering glucose variability would lower the rate of late-onset preeclampsia. The cohort is from a specific population, and the effect sizes for the mediated pathway, while real, are modest. The authors themselves frame variability as a potential mediating factor and call for future preventive strategies to be tested.
So no, this is not a green light to buy a continuous glucose monitor in early pregnancy on your own initiative, or to start aggressively cutting carbs without your obstetric team in the loop. Pregnancy nutrition has real guardrails — caloric needs, micronutrients, ketone considerations — and DIY metabolic experimentation isn't the move. What this study does justify is a sharper conversation with your clinician, especially if you're entering pregnancy with a higher BMI or a history of metabolic issues.

Movement, sleep and meal composition are the everyday inputs that influence glucose curves — and the ones a care team can help personalize.
The bigger picture for metabolic-health readers
If you've been following the broader metabolic-health conversation — CGMs, time-in-range, the gospel of stable glucose — this study lands in a familiar place with an unfamiliar audience. Pregnancy research has historically been conservative about importing trendy metabolic concepts, and rightly so: the physiology is different, the stakes are higher, and the evidence base is thinner. The fact that a peer-reviewed multicenter analysis is now using structural equation modeling to map glucose variability as a mediator of a serious pregnancy outcome is a sign the field is starting to take the question seriously.
The takeaway isn't a hack. It's a reframing. Pre-conception is metabolic prep, not just folic acid and prenatal vitamins. Early pregnancy is a window where the body's glucose handling may be quietly telling a story about what's coming in the third trimester. And late-onset preeclampsia — long considered nearly impossible to predict and even harder to prevent — may be slightly less mysterious than it used to be.
Moderate evidence, real signal, no miracle. That's the honest headline.
Pre-conception is metabolic prep — not just folic acid and prenatal vitamins.
Frequently asked questions
What did the study find about glucose variability and late-pregnancy blood pressure problems?
In 802 pregnancies across 14 centers in Japan, researchers found that high glucose variability in early pregnancy partially mediated the link between higher pre-pregnancy BMI and late-onset hypertensive disorders of pregnancy. Overweight and obese participants had high glucose variability at a rate of 26.1% versus 16.4% in non-overweight participants, and late-onset hypertensive disorders at 17.6% versus 7.9%.
If I pass the standard gestational diabetes screening, does that mean glucose variability isn't a concern for me?
Not necessarily. The study found that the association between glucose variability and late-onset hypertensive disorders was actually stronger in pregnancies without gestational diabetes than in the overall group. The article notes this challenges the binary idea that you either have GDM or you don't — you can pass the standard screen and still carry a metabolic signal that may be relevant to later blood-pressure complications.
Does this study mean I should buy a continuous glucose monitor during early pregnancy?
The article explicitly advises against acting on this research on your own. Because the study is a retrospective observational analysis rather than a randomized trial, it does not prove that lowering glucose variability would lower the rate of late-onset preeclampsia, and the article cautions against purchasing a CGM or aggressively cutting carbs without your obstetric team involved.
How is glucose variability different from the blood sugar numbers most people already track?
Most blood sugar measures — like A1C, fasting glucose, or the OGTT pass/fail result — reflect an average, while glucose variability looks at how jagged the curve is across the day, including the peaks after meals and the dips between them. Outside of pregnancy, glucose variability has been linked in observational research to vascular stress, oxidative damage, and endothelial dysfunction, which are the same biological areas where preeclampsia develops.
What are the main limitations of this research that readers should keep in mind?
The study is a retrospective observational analysis, not a randomized trial, so the structural equation modeling describes statistical relationships in a snapshot of data rather than proving that any intervention would change outcomes. The cohort comes from a specific population, the effect sizes for the mediated pathway are described as modest, and the authors themselves call for future preventive strategies to be tested.
Sources

The Sirtuin–Senescence Axis: Mapping the Molecular Drivers of Aging
Three converging 2025 reviews sketch an integrated picture of why tissues deteriorate—and where senolytics, NAD+ modulators, and lysosome-restoring therapies might one day intervene.
Aging is finally beginning to look less like a vague decline and more like a wiring diagram. Across three converging 2025 reviews and primary studies, a coherent story is taking shape: the enzymes that maintain cellular housekeeping run down, the cells that should retire instead linger and inflame their neighbors, and the tiny acidic compartments responsible for clearing molecular debris quietly lose their charge. None of these threads is new on its own. What's notable now is how tightly they braid together—and how many of the resulting targets are already in early clinical trials.
Three threads, one fabric
The first thread runs through the sirtuins, a family of seven NAD+-dependent enzymes (SIRT1–SIRT7) that act as cellular stress sensors. A recent review in Pharmaceuticals traces how sirtuins govern metabolism, DNA repair, and the stress response across the female lifespan, and how their decline with age contributes to menopausal and metabolic complications, with NAD+ precursors and SIRT1 activators emerging as plausible interventions. The framing matters: sirtuins are not a youth switch but a network whose isoform- and tissue-specific roles are still being mapped.
The second thread is cellular senescence—the state in which a damaged cell stops dividing but refuses to die, instead secreting a cocktail of inflammatory signals known as the senescence-associated secretory phenotype, or SASP. A comprehensive 2025 review in the International Journal of Molecular Sciences argues that senescent cells are not a single population but a heterogeneous mix that varies by tissue and disease context, and identifies p53/p21, p16INK4a/RB, mTOR, and p38 MAPK as the core pathways driving senescence and SASP production in lung aging.
The third thread is the quietest and perhaps most mechanistically satisfying. Lysosomes—the acidic compartments that recycle worn-out proteins—depend on a molecular pump called V-ATPase to maintain their low pH. In a yeast replicative-aging model published in Aging Cell, researchers showed that V-ATPases physically disassemble into their V1 and V0 subcomplexes in older cells, alkalinizing the vacuole and degrading lysosomal function. Caloric restriction, a long-standing lifespan extender across species, prevented that disassembly and preserved vacuolar pH.

Lysosomes are the cell's recycling centers; when their internal pH drifts, the entire waste-clearance system stutters.
Aging is finally beginning to look less like a vague decline and more like a wiring diagram.
Why these three threads belong in one story
Read in isolation, each paper is a specialist's report. Read together, they describe a feedback loop. Sirtuins depend on NAD+, a coenzyme whose availability falls with age. As sirtuin activity drops, the stress responses that keep damaged cells from tipping into senescence weaken. Senescent cells then accumulate and broadcast SASP signals that further disrupt metabolism in neighboring tissues. Meanwhile, the lysosomes those neighbors rely on to clear damaged proteins are themselves losing acidity, leaving cells less able to dispose of the very debris that triggers senescence in the first place.
The Pharmaceuticals review makes the case that sirtuins sit at the intersection of reproductive function, hormone-dependent cancers, and age-related metabolic disease—a hub, not a single lever. The lung-focused review extends the logic to tissue-level pathology, noting that accumulated senescent cells help drive lung age-related diseases, and that clearing or quieting them has shown promise in preclinical and early clinical work.
What the drug pipeline actually looks like
The therapeutic vocabulary here splits cleanly. Senolytics selectively trigger apoptosis in senescent cells. Senomorphics (or senostatics) leave the cells in place but quiet their inflammatory secretions. The IJMS review catalogs both camps: natural compounds such as quercetin, fisetin, and resveratrol, alongside repurposed drugs including dasatinib, navitoclax, metformin, and rapamycin, are in clinical trials for age-related lung disease and broader healthspan endpoints. The same review is candid that senescent-cell heterogeneity complicates the picture—different tissues harbor different subpopulations, and a senolytic that clears one may miss another.
On the sirtuin side, the most discussed interventions are NAD+ precursors and direct SIRT1 activators. The Pharmaceuticals review frames these as promising in mitigating menopausal and metabolic complications—language that, importantly, stops well short of proven clinical benefit at the population level. The lysosomal story is earlier still: the V-ATPase work is mechanistic and performed in yeast, and while the team identified the RAVE complex and Oxr1 as opposing regulators of V-ATPase assembly, with Rav2 overexpression delaying disassembly and extending replicative lifespan, translating that into a human therapeutic is years of work away.

Quercetin and fisetin—two of the most-studied natural senolytic candidates—are abundant in common produce, though dietary intake is not equivalent to the doses used in trials.
- One axis, three layers. Sirtuin decline, senescent-cell accumulation, and lysosomal acidification loss appear to reinforce each other rather than act independently.
- Senolytics vs. senomorphics. Two distinct strategies—killing senescent cells or quieting their secretions—are both in clinical testing for age-related disease.
- NAD+ matters, but isn't magic. Sirtuin-supporting interventions show preclinical promise; durable human outcomes are not yet established.
- Caloric restriction still earns its keep. In yeast, it directly prevented the V-ATPase disassembly that degrades lysosomal function with age.
- Tissue heterogeneity is the catch. Senescent cells differ by organ and disease, so a single drug is unlikely to be universally effective.
- This is a map, not a manual. The evidence supports the framework more strongly than it supports any specific intervention.
The honest read
What makes this moment interesting is not that any one paper is definitive—none is—but that the three frames are converging on a shared causal architecture. The hallmarks-of-aging research program has long been criticized for cataloging phenomena without mechanistically linking them. The sirtuin–senescence–lysosome axis is one of the cleaner attempts at exactly that linkage, and it is producing testable predictions: if you restore NAD+, you should bias cells away from senescence; if you clear senescent cells, you should reduce SASP-driven dysfunction in surrounding tissue; if you stabilize V-ATPase assembly, you should slow at least one component of lysosomal aging.
None of those predictions has been confirmed in a definitive human trial. Many of the candidate drugs—rapamycin, metformin, navitoclax—carry real side-effect profiles that make casual use unwise. The credible posture for a longevity-curious reader is the same one the field's better scientists hold: the framework is increasingly coherent, the targets are increasingly specific, and the clinical proof is still pending. That's a meaningful upgrade from where this field sat even five years ago—and it is not, yet, a permission slip.
Frequently asked questions
What are sirtuins, and why does their decline matter for aging?
Sirtuins are a family of seven NAD+-dependent enzymes (SIRT1–SIRT7) that act as cellular stress sensors, governing metabolism, DNA repair, and the stress response. Their activity falls with age, and that decline contributes to menopausal and metabolic complications. The article describes them not as a single youth switch but as a network whose isoform- and tissue-specific roles are still being mapped.
What is the SASP, and why is it a problem?
SASP stands for senescence-associated secretory phenotype—a cocktail of inflammatory signals secreted by senescent cells, which are damaged cells that stop dividing but refuse to die. Those signals further disrupt metabolism in neighboring tissues, and accumulated senescent cells are described as helping drive lung age-related diseases.
What is the difference between a senolytic and a senomorphic?
Senolytics selectively trigger apoptosis in senescent cells, whereas senomorphics—also called senostatics—leave the cells in place but quiet their inflammatory secretions. Both strategies are currently being tested in clinical trials for age-related disease.
How does caloric restriction connect to lysosomal function?
Lysosomes rely on a molecular pump called V-ATPase to maintain the low internal pH that allows them to recycle worn-out proteins. In a yeast aging model, V-ATPases physically disassembled in older cells, raising the vacuolar pH and degrading lysosomal function. Caloric restriction prevented that disassembly and preserved vacuolar acidity.
Which natural compounds are being studied as senolytic candidates?
The article lists quercetin, fisetin, and resveratrol as natural compounds in clinical trials for age-related lung disease and broader healthspan endpoints. Quercetin and fisetin are specifically described as two of the most-studied natural senolytic candidates and are noted to be abundant in common produce.
Sources
- Sirtuins in Women's Health. — Pharmaceuticals (Basel, Switzerland)
- Heterogeneity of Cellular Senescence, Senotyping, and Targeting by Senolytics and Senomorphics in Lung Diseases. — International journal of molecular sciences
- V-ATPase Disassembly at the Yeast Lysosome-Like Vacuole Is a Phenotypic Driver of Lysosome Dysfunction in Replicative Aging. — Aging cell

Mito-Inhibitors in the Bloodstream: A New Clue to Alzheimer's Bioenergetic Decline
A small serum study fingers two circulating lipids — nervonic acid and 15-epi-PGA1 — that throttle mitochondria in a dish. It's early, but it reframes Alzheimer's as a whole-body energy problem.
For two decades, the story of Alzheimer's disease has been told largely above the neck — plaques and tangles, misfolded proteins, a brain quietly betraying itself. But a quieter subplot has been gathering evidence: mitochondria, the cell's power plants, sputter throughout the body in people with Alzheimer's, not just in their neurons. The obvious question — why? — has been harder to answer. A new study in GeroScience offers a provocative, still-early clue: maybe the brain isn't the only thing sabotaging itself. Maybe the blood is carrying the saboteurs.
The paper, led by Heimler and colleagues, took serum from older adults across three groups — cognitively normal, mild cognitive impairment, and dementia — and ran it through mass spectrometry to hunt for lipid metabolites that might directly suppress mitochondrial function. The team layered three filters: did the molecule's abundance track with how donor serum affected energy production in naïve cells in a dish; did it track with the bioenergetic capacity of the donor's own blood cells; and did it track with cognitive scores on the modified mini-mental state exam. Two molecules survived all three filters: nervonic acid and 15-epi-Prostaglandin A1 (15-epi-PGA1), both elevated in participants with dementia compared with cognitively normal peers.
What the lipids actually did in a dish
Identifying a suspicious molecule in serum is one thing; showing it can throttle a mitochondrion is another. The investigators applied the candidate lipids to neurons, myoblasts, and fibroblasts, then used high-resolution respirometry — essentially, measuring how fast cells consume oxygen under controlled metabolic challenges — to see what happened. Both nervonic acid and 15-epi-PGA1 inhibited mitochondrial function across all three cell types, acting through broad suppression of the electron transfer system without changing overall mitochondrial content.
That last detail matters. The cells weren't losing mitochondria; the mitochondria they had were simply working less efficiently. It's the difference between a power plant being demolished and a power plant being told to throttle back. If the finding generalizes, it suggests a mechanism that could quietly drag down energy production in tissues far from any amyloid plaque.

Respirometry experiments measured how cells breathe when exposed to the candidate lipids.
The cells weren't losing mitochondria. The mitochondria they had were simply working less efficiently.
Why this reframes the bioenergetic story
For years, researchers have noted that people with Alzheimer's show signs of impaired energy metabolism in cells that have nothing to do with the brain — skin fibroblasts, blood cells, muscle. The standard explanation has been that some shared genetic or aging-related vulnerability shows up everywhere at once. This study floats a different possibility, framed cautiously by the authors themselves: that circulating molecules in serum may actively drive systemic bioenergetic decline in the context of Alzheimer's dementia. Not just a passive marker of disease, but a possible participant.
If that holds up, it changes the kind of intervention worth thinking about. Brain-targeted therapies are hard. Modulating a circulating lipid pool — through diet, metabolism, or pharmacology — is, at least in principle, a more tractable problem. But that's a long bridge from a serum sample and a Seahorse plate.
What's worth holding onto, and what isn't
It would be easy to mistranslate this finding into supplement-aisle advice. Nervonic acid, in particular, has been marketed for years as a brain-support ingredient, often in fish-oil-adjacent blends. Nothing in this study endorses any consumer product, and the direction of the finding is, if anything, the opposite of the marketing pitch: the lipid was elevated in dementia and suppressed mitochondrial function in cells. That does not mean dietary nervonic acid causes Alzheimer's — circulating levels reflect a complex stew of synthesis, diet, and turnover — but it should temper any confident claim that more of it is good for your brain.
15-epi-PGA1, a prostaglandin derivative, is not on shelves and is unlikely to be. Its interest here is purely mechanistic: a clue about which biochemical pathways might be tilting the wrong way in dementia.

- Two suspects, not a verdict. Nervonic acid and 15-epi-PGA1 were elevated in dementia serum and inhibited mitochondrial function across three cell types in vitro.
- Mechanism, not magnitude. The lipids broadly suppressed the electron transfer system without reducing mitochondrial number — a throttling effect, not a destructive one.
- Systemic, not just neural. The findings support a model in which Alzheimer's bioenergetic decline is partly driven by molecules circulating through the body.
- Early evidence. A single serum-plus-in-vitro study cannot establish that altering these lipids would help patients; replication and human trials are needed.
- Not a shopping list. Nothing here justifies starting — or stopping — any supplement; talk to a clinician about cognitive health concerns.
The honest read on this paper is that it is a thoughtful first map of a territory that hasn't been charted carefully before. The authors did the unglamorous work of triangulating correlations across serum effects, donor cells, and cognition before declaring a candidate. That discipline is exactly what an early finding in Alzheimer's needs, because the field has been burned, repeatedly, by bold claims that didn't survive replication.
For now, the useful posture is curiosity, not certainty. If circulating lipids really are part of how Alzheimer's drains the body's energy supply, the next few years of follow-up work will tell us. Until then, the most evidence-based thing a reader can do for their mitochondria is the boring, durable stuff: sleep, movement, cardiovascular health, and a conversation with a clinician about any cognitive changes worth tracking.
Frequently asked questions
What are nervonic acid and 15-epi-PGA1, and why did this study flag them?
Both are lipid metabolites found at elevated levels in the blood serum of people with dementia compared with cognitively normal participants. They were the two molecules to survive all three filters the researchers applied: their abundance tracked with how donor serum affected energy production in cells, with the bioenergetic capacity of donors' own blood cells, and with cognitive test scores.
How exactly did these lipids affect mitochondria in the laboratory?
When applied to neurons, myoblasts, and fibroblasts, both molecules inhibited mitochondrial function across all three cell types by broadly suppressing the electron transfer system. Critically, the cells were not losing mitochondria — the mitochondria they had were simply working less efficiently, a throttling effect rather than a destructive one.
Does this study mean people should avoid nervonic acid supplements?
The article states that nothing in this study endorses or condemns any consumer product, and it cautions that the direction of the finding is the opposite of typical marketing claims — nervonic acid was elevated in dementia and suppressed mitochondrial function in cells. It also notes that circulating levels of the lipid reflect a complex mix of synthesis, diet, and turnover, so the finding does not mean dietary nervonic acid causes Alzheimer's.
Does the study prove that these lipids cause Alzheimer's disease?
No. The authors themselves frame their conclusion cautiously: this is a single mechanistic study that identifies a correlation and a plausible mechanism, but it does not establish causation in humans. Replication in larger, independent cohorts and eventually in animals and clinical trials would be needed before the finding could be considered actionable.
Why is it significant that the mitochondrial effects were seen outside the brain?
Researchers have long noted that people with Alzheimer's show signs of impaired energy metabolism in cells unrelated to the brain, such as skin fibroblasts, blood cells, and muscle, typically attributed to a shared genetic or aging-related vulnerability. This study raises a different possibility: that molecules circulating in the blood may actively drive that systemic energy decline, making them a potential target distinct from brain-focused therapies.
Sources

Inside the Army's Bid to Put Whole-Person Health on a Single Dashboard
The US Army's new Holistic Health and Fitness Management System tries to unify sleep, nutrition, biometrics and mental health for hundreds of thousands of soldiers. It's the largest real-world test yet of population-scale optimization.
For the past decade, the language of personal optimization has belonged to consumers — the executive with a ring on her finger, the founder tracking heart-rate variability before a board meeting, the coach quietly titrating magnesium and morning light. Now the same logic is being stress-tested at a scale the wellness industry has never approached: the entire active US Army. A 2025 review in BMJ Military Health describes the Holistic Health and Fitness Management System, or H2FMS, a cloud-based platform built to assess, coach and track soldiers across five readiness domains at once — physical, mental, spiritual, nutritional and sleep. For anyone who has ever wondered what 'whole-person' health looks like when it is funded, mandated and operationalised, this is the closest real-world answer we have.
- One dashboard, five domains. H2FMS unifies physical, mental, spiritual, nutritional and sleep data into a single role-based view.
- Population scale, not personal. It is designed for hundreds of thousands of soldiers and the interdisciplinary teams that support them — not a quantified-self individual.
- Inputs are mixed. Biometric wearables sit alongside self-reported psychometrics, performance tests and legacy Army systems.
- Evidence is architectural, not outcome-based yet. The published review describes design and intent; population-level health outcomes have not yet been reported.
- The signal for civilians. Integration — not more sensors — is becoming the frontier of credible health technology.
Why the Army stopped thinking in silos
For most of modern military history, soldier readiness was tracked the way corporate wellness is still tracked today: in disconnected silos. A physical fitness test lived in one system. Sleep complaints surfaced — if at all — in a clinic note. Nutrition was a dining-facility problem. Mental health sat behind its own door, with its own stigma and its own records. The premise of H2FMS, according to the BMJ Military Health review, is that those silos themselves are the problem: a soldier whose sleep is collapsing is also the soldier whose injury risk is climbing and whose cognitive performance is degrading, and no one downstream can see the whole picture in time to intervene.
The platform's response is structural. The authors describe a secure, cloud-based system that pulls together advanced biometric technologies, performance assessments, self-reported psychometrics and existing Army systems, then surfaces them through role-based dashboards — one view for the individual soldier, another for the Holistic Health and Fitness Performance Team, another for cadre and command. The point is not to give anyone more data. It is to give the right person the right slice, at the moment a decision is being made.

H2FMS combines wearable biometrics with self-reported inputs — an acknowledgement that no single signal captures readiness.
What 'holistic' actually means here
The word holistic has been so thoroughly laundered by the wellness market that it is worth pausing on what the Army means by it. The review defines five readiness domains: physical, mental, spiritual, nutritional and sleep. Spiritual readiness is the one that tends to surprise civilian readers — in the Army's framing it is closer to a sense of meaning, values and purpose than to religious practice, and it is treated as a measurable contributor to resilience alongside VO2 max and sleep efficiency.
What ties the domains together inside H2FMS is not a single score but a set of mechanisms: automated support actions that trigger when a metric drifts, domain-specific tools for clinicians and coaches, guided education and interactive coaching for the soldier, and integrated analytics that let teams allocate finite resources where the data says they will matter most. The platform is explicitly designed, the authors write, to enable data-informed interventions intended to boost performance, reduce injuries, speed rehabilitation and improve quality of service.
The frontier is no longer more sensors. It is whether anyone can act on what the sensors already say.
Population scale changes the math
Consumer optimization is a story of one person and their data. Military readiness is a story of resource allocation across a population that is too large to coach individually. The H2FMS authors describe the platform as a cost-efficient strategy for scaling Holistic Health and Fitness Performance Team access across the Total Army, framed as an equitable model meant to maximise both individual and organisational return on investment. In plainer language: every soldier gets on-demand access to expert-informed support, and the limited supply of human coaches and clinicians is steered toward the soldiers whose data suggests they need it most.
That logic has obvious parallels in civilian life. The most expensive parts of any health system — specialists, intensive rehab, time — are scarce. Platforms that triage attention based on integrated signals, rather than waiting for a complaint or a crisis, are precisely what large employers and health systems have been promising for a decade. H2FMS is one of the first credible attempts to actually build that architecture for a defined population at this scale.

The dashboard is meant for teams, not just individuals — the platform's leverage comes from coordinating specialists around the same view.
What the evidence does — and does not — say
This is where editorial honesty matters. The BMJ Military Health piece is a review of a system's architecture and intent, not a randomised trial of its effects. It describes what H2FMS is built to do — unify data, trigger support, inform decisions — and the rationale for why that design should improve outcomes. It does not yet report population-level evidence that injuries fell, that rehabilitation accelerated, or that quality of service measurably improved as a result of the platform. Those are the outcomes the system is designed to produce, and they remain to be demonstrated in published data.
That distinction is the whole game for readers trying to separate signal from hype in wellness technology. A platform can be well-architected and still under-deliver in the field; it can also quietly compound small gains across a population in ways that take years to surface in the literature. The honest read on H2FMS today is that it is a serious, well-specified bet on integration as the next lever in human performance — and that the proof will arrive, or fail to arrive, in the outcome studies that follow.
The signal for the rest of us
For PinnacleLife readers, the interesting thing about H2FMS is not that the Army is using wearables. It is that a large, conservative institution has concluded that the bottleneck in human performance is no longer measurement. It is the connective tissue between measurement, decision and action — and that connective tissue has to be built deliberately, with role-based views, automated triggers and humans in the loop. That is a quietly radical position in a market that still mostly sells more sensors.
If the platform works as designed, the lesson for the rest of us will be familiar but worth relearning: the best health technology is the kind that compresses the distance between a signal and a sensible response. If it does not work as designed, the lesson will be just as useful — that even with budget, mandate and scale, 'whole-person' optimization is harder to operationalise than the wellness industry likes to suggest. Either way, H2FMS is the experiment to watch.
Frequently asked questions
What are the five health domains tracked by H2FMS?
H2FMS tracks soldiers across five readiness domains: physical, mental, spiritual, nutritional, and sleep. These are unified into a single cloud-based platform accessible through role-based dashboards.
What does the Army mean by 'spiritual readiness' — is it about religion?
In the Army's framing, spiritual readiness is closer to a sense of meaning, values, and purpose than to religious practice. It is treated as a measurable contributor to resilience alongside metrics like VO2 max and sleep efficiency.
Has H2FMS been shown to reduce injuries or improve soldier health outcomes?
Not yet in published data. The BMJ Military Health review describes the system's architecture and intent, not a randomized trial of its effects. Population-level evidence that injuries fell, rehabilitation accelerated, or quality of service improved has not yet been reported.
Who can see a soldier's data on the platform?
H2FMS uses role-based dashboards, giving different views to the individual soldier, the Holistic Health and Fitness Performance Team, and cadre and command. The design goal is to give the right person the right slice of data at the moment a decision is being made.
How does H2FMS decide which soldiers receive the most attention from coaches and clinicians?
The platform uses integrated analytics so that the limited supply of human coaches and clinicians is steered toward the soldiers whose data suggests they need it most. The authors describe this as a cost-efficient, equitable model intended to maximize both individual and organizational return on investment.
Sources

The New Peptide Pipeline: How AI Is Quietly Rewiring Drug Discovery
Computational design is pushing peptide therapeutics into oncology, infectious disease, and antimicrobial resistance. The early signals are real — and the caveats matter.
Peptides have been having a long moment. GLP-1 drugs put them on magazine covers; gym-floor chatter put fragments like BPC-157 into group chats. But the more interesting story is happening upstream of the hype: a wave of computational tools — topology-enhanced machine learning, reactive-force-field molecular dynamics, AI-driven repurposing — is starting to design and refine therapeutic peptides faster than wet labs alone ever could. The headline for a busy 40-year-old isn't that peptides will solve your Monday. It's that the pipeline behind the next decade of targeted therapies is being rebuilt, quietly, in silico.
Therapeutic peptides sit in a useful middle ground between small-molecule drugs and full antibodies: specific enough to hit a single target, small enough to be engineered at scale, and — increasingly — tractable for AI models that can screen millions of candidate sequences before a single one is synthesized. Across five recent reviews and methods papers, the same throughline keeps surfacing: computational design is becoming the engine, and peptides are the payload.
- Computation is moving upstream. AI and molecular-dynamics tools are now shaping which peptides get made, not just analyzing what's already in the freezer.
- Oncology is the proving ground. Peptides targeting HER2, VEGF, and EGFR are being explored as more selective alternatives or complements to conventional chemotherapy.
- Antimicrobial peptides are a credible answer to resistance — in theory. Their multi-pronged mechanisms make resistance harder to evolve, but most data are preclinical.
- Antivirals are getting smaller and sharper. A redesigned HIV-1 entry inhibitor was cut in half and reportedly gained over 100-fold potency in lab assays.
- Stability and cost are still the bottleneck. Enzymatic degradation, bioavailability, and manufacturing remain the unsexy problems gating real-world use.
Oncology: smarter targeting, fewer collateral hits
The pitch for peptides in cancer therapy is precision. A 2025 review argues that multifunctional peptides offer high specificity, minimized toxicity, and the ability to influence multiple pathways at once — including HER2, VEGF, and EGFR, three of the most consequential signaling axes in breast cancer. The same review describes peptide-based vaccines, immune-modulating sequences, and peptide-drug conjugates designed to drag chemotherapy payloads directly to tumor tissue while sparing healthy cells.
That's the promise. The honest reading is that most of this work is still in preclinical or early-clinical territory, and the authors are blunt about the obstacles: enzymatic degradation, limited stability, and high production costs remain the practical drag on the field. Engineering tricks — cyclization, stapling, cell-penetrating modifications — are the workarounds being tested.

Computational screening lets researchers triage thousands of peptide candidates before committing to synthesis.
The machine-learning layer
The reason any of this is moving faster than it did a decade ago is that the screening bottleneck is loosening. A new method called Top-ML uses topological features derived from a peptide's sequence "connection" information to predict anticancer activity — and reportedly matches state-of-the-art deep learning performance on standard benchmark datasets while being more interpretable about why it picked a given candidate.
Interpretability matters more than it sounds. A model that flags a promising sequence and can explain which structural features drove the call is a model a medicinal chemist can actually act on. That's the quiet shift: from black-box screening to design tools that argue their reasoning.
The bottleneck used to be which peptides to make. Increasingly, it's which ones to believe.
Antivirals: shrinking the molecule, sharpening the hit
The most striking single result in this batch comes from HIV research. A team used ReaxFF molecular dynamics — a reactive-force-field simulation method — to dissect how each amino acid in VIRIP, a 20-residue natural fragment of α1-antitrypsin, contributes to blocking the HIV-1 gp41 fusion peptide. By trimming the peptide based on that analysis, they produced a 10-residue version (soVIRIP) with more than 100-fold higher antiviral activity than the original and an IC50 around 120 nM in infection assays. It was also reported as nontoxic in a zebrafish model.
The context for that number: a dimeric VIRIP derivative (VIR-576) has already cleared a phase I/II clinical trial for safety and efficacy, so this isn't a from-scratch program — it's an optimization of a peptide that already has human data behind it. Two things are notable for a non-clinician reader. First, smaller peptides are cheaper to manufacture and easier to formulate. Second, the design move was made in silico, then validated in the lab — the inverse of the usual workflow.
The same computational-repurposing logic is being applied to SARS-CoV-2. A 2025 review argues that AI-driven mass screening can support a peptide repurposing strategy analogous to small-molecule repurposing, identifying existing therapeutic peptides that might modulate immune responses, block viral entry, or disrupt replication. Stability, bioavailability, and viral mutation remain the open problems.
Antimicrobial peptides and the resistance problem
AMPs disrupt bacterial membranes directly — a mechanism that's harder for pathogens to evolve around than single-target antibiotics.
Antibiotic resistance is the slow-motion crisis that should be on every health-literate adult's radar. Antimicrobial peptides (AMPs) — small, often cationic sequences produced by the innate immune systems of organisms from frogs to humans — are one of the more credible candidates to back up a thinning antibiotic arsenal.
A 2025 review summarizes why: AMPs can permeabilize bacterial membranes, limit biofilm formation, and modulate immune responses, attacking pathogens on multiple fronts at once. That multi-mechanism profile is what makes resistance harder to evolve than against a conventional antibiotic that hits a single enzyme. The same review notes activity reported across bacteria, viruses, fungi, and cancer cells.
The caveats are the usual ones for this field: most of the strongest data are preclinical, clinical translation has been slow, and turning a promising AMP into a stable, affordable drug is still hard. The direction of travel is encouraging; the timeline for a pharmacy-shelf AMP is not next year.
What to actually take from this
The evidence rating here is moderate, and the language should match. Across oncology, antivirals, and antimicrobials, peptides plus computation look like a genuinely productive pairing — strong enough to take seriously, early enough that overclaiming would be dishonest. The most defensible read for a non-clinician: the design tools are real, some of the lab results are striking, and the translation to approved human therapies will take years and will not look like the consumer-peptide marketplace currently looks.
For now, the practical move is to update your mental model of where drug discovery is heading, not your supplement stack. The pipeline is being rebuilt. The shelf hasn't caught up yet.
Frequently asked questions
What makes therapeutic peptides different from other types of drugs?
Therapeutic peptides sit in a middle ground between small-molecule drugs and full antibodies: they are specific enough to hit a single target and small enough to be engineered at scale. They are also increasingly tractable for AI models that can screen millions of candidate sequences before a single one is synthesized.
What happened when researchers redesigned the HIV-fighting peptide VIRIP?
Using ReaxFF molecular dynamics to analyze each amino acid's contribution, researchers trimmed the original 20-residue peptide to a 10-residue version called soVIRIP, which showed more than 100-fold higher antiviral activity than the original and an IC50 of around 120 nM in infection assays. It was also reported as nontoxic in a zebrafish model.
Why are antimicrobial peptides considered a credible response to antibiotic resistance?
Antimicrobial peptides can permeabilize bacterial membranes, limit biofilm formation, and modulate immune responses, attacking pathogens on multiple fronts simultaneously. That multi-mechanism profile makes resistance harder to evolve than against a conventional antibiotic that hits a single enzyme.
What is Top-ML and why does the article highlight its interpretability?
Top-ML is a method that uses topological features derived from a peptide's sequence connection information to predict anticancer activity, reportedly matching state-of-the-art deep learning performance on standard benchmarks. Its interpretability matters because a model that can explain which structural features drove its prediction gives medicinal chemists something they can actually act on, unlike a black-box screening tool.
What are the main obstacles still preventing peptide therapies from reaching patients?
The article identifies enzymatic degradation, limited stability, poor bioavailability, and high production costs as the key practical barriers. Engineering approaches such as cyclization, stapling, and cell-penetrating modifications are among the workarounds currently being tested.
Sources
- Peptides in breast cancer therapy: From mechanisms to emerging drug delivery and immunotherapy strategies. — Pathology, research and practice
- Topology-Enhanced Machine Learning Model (Top-ML) for Anticancer Peptide Prediction. — Journal of chemical information and modeling
- ReaxFF-Guided Optimization of VIRIP-Based HIV-1 Entry Inhibitors. — The journal of physical chemistry. B
- Rerouting therapeutic peptides and unlocking their potential against SARS-CoV2. — 3 Biotech
- Antimicrobial peptides: a promising frontier to combat antibiotic resistant pathogens. — Annals of medicine and surgery (2012)
GLP-1s Beyond Glucose: Liver, Plaque, and a Pancreas Warning Shot
Semaglutide, exendin-4, and tirzepatide are pushing GLP-1 receptor agonists into hepatology and cardiology — while a fresh case report reminds us the safety ledger is still open.
The GLP-1 receptor agonist story used to be tidy. A gut peptide mimicked in the lab, deployed against type 2 diabetes, repurposed for weight loss, and championed in cardiometabolic clinics. Tidy is over. In the span of a few months, three new papers have pushed semaglutide into hepatology, exendin-4 into vascular immunology, and tirzepatide into a cautionary case file. None of it overturns the existing playbook. All of it suggests the playbook is about to get longer — and that the quantified-self crowd already micro-dosing these molecules should be reading the fine print, not just the headlines.
- Liver signal, rodent only. Semaglutide blunted chemotherapy-induced hepatotoxicity in rats via PINK1/Parkin mitophagy and NF-κB/NLRP3 suppression — mechanism-rich, but not yet human data.
- Plaque biology, bench only. Exendin-4 reduced foam-cell formation and inflammation in macrophages and ApoE-/- mice by modulating a TREM2–JAK2/STAT3 axis.
- Pancreatitis, single case. A 32-year-old developed probable acute pancreatitis five weeks into tirzepatide, with lipase above 11,000 U/L.
- Evidence rating: Early. Preclinical mechanism plus an n-of-1 adverse event is hypothesis-generating, not prescriptive.
- Talk to a clinician. Anyone using or considering these peptides should be having this conversation with a prescribing physician, not a forum.
The liver experiment that wasn't about glucose
The first of the three papers takes semaglutide somewhere it isn't usually asked to go: the chemotherapy ward. 5-fluorouracil (5-FU) is a workhorse cytotoxic that comes with well-documented hepatic collateral damage. A research group dosed rats with 5-FU, then pretreated a subset with oral semaglutide, and looked at what happened inside the hepatocytes.
The protective signal was substantial. Pretreated animals showed lower hepatic enzymes, less oxidative stress, fewer inflammatory markers, and cleaner histology than 5-FU controls. The mechanism the authors propose is the interesting part: semaglutide appeared to enhance PINK1/Parkin-mediated mitophagy while suppressing the ROS/NF-κB/NLRP3 inflammasome axis, with proinflammatory cytokines TNF-α and IL-6, the oxidative marker malondialdehyde, and apoptotic caspase signaling all trending down. Co-administering chloroquine — an autophagy inhibitor — abolished the benefit, which is the kind of mechanistic clincher reviewers like to see.
The caveat is the one any careful reader has already supplied. This is a rat model of a specific chemotherapy injury, not a human trial of fatty-liver disease, not a real-world signal in oncology patients, and not a license to add semaglutide to a chemo regimen. It is, however, a credible mechanistic claim for hepatoprotection that someone will now try to replicate in larger animals and, eventually, people.
The semaglutide hepatoprotection signal lives in rodent histology — promising, preclinical, and a long way from a human indication.
Exendin-4 walks into the plaque
The second paper concerns exendin-4, the lizard-venom-derived peptide that gave the GLP-1 class its first foothold. Cardiovascular benefit from GLP-1 agonism is one of the better-substantiated extra-glycemic effects, but the cellular wiring has remained murky. Here, a Chinese group zoomed in on macrophages, the cells that gulp oxidized LDL and turn into the foam cells that build arterial plaque.
Stimulating THP-1-derived macrophages with oxLDL ramped up expression of TREM2, an immunoreceptor with a controversial role in atherosclerosis, and activated downstream JAK2/STAT3 signaling — a combination that accelerated foam-cell formation and pumped out inflammatory cytokines. Exendin-4 intervened on that circuit. In both the cell model and ApoE-/- mice on a high-fat diet, the peptide attenuated foam-cell formation and inflammation by regulating the TREM2–JAK2/STAT3 axis.
This is the kind of paper that reframes a known clinical effect rather than discovering a new one. We already suspected GLP-1 agonists do something useful inside arteries; this work suggests one of the levers is immunometabolic, sitting on a receptor that researchers are still arguing about. The translational distance to a human plaque-stabilization claim remains long, and the cohort is a mouse model — but mechanism matters when you're trying to predict which patients benefit most.
Mechanism is not a clinical claim — but it tells you where to look for one. PinnacleLife
A pancreas, a five-week timeline, and a number worth remembering
The third paper is the shortest and, for readers who actually use these drugs, the loudest. A 32-year-old woman with a prior history of gestational diabetes started tirzepatide for weight loss. Four weekly doses at 2.5 mg, then a single 5 mg dose. Five weeks in, she presented with three weeks of worsening epigastric pain, nausea, vomiting, and constipation. Her lipase came back at 11,645 U/L — multiple times the upper limit of normal. Imaging confirmed acute interstitial oedematous pancreatitis, with incidental gallstones but no biliary obstruction on MRCP.
The clinicians stopped the tirzepatide, managed conservatively, and watched her lipase normalize by discharge. They classified this as a probable case of tirzepatide-induced acute pancreatitis, using standard causality reasoning given the temporal relationship and absence of an alternative culprit despite the gallstones.
A single case report is exactly what it sounds like: one data point. The GLP-1 / pancreatitis question has been litigated for over a decade, and large datasets have generally found the absolute risk small. But tirzepatide is a newer molecule — a dual GLP-1/GIP agonist — and the post-marketing surveillance picture for it is still being filled in. For a quantified-self reader, the practical takeaway is unglamorous: persistent upper abdominal pain on any incretin-class agent is a reason to stop dosing and get a lipase, not to wait it out.

Lipase is the cheap, fast test that turns abdominal pain on a GLP-1 from a guessing game into a decision.
What an evidence-literate reader does with this
Stitched together, these three papers describe a class that is doing more than lowering A1c and shrinking waistlines. The hepatology and vascular biology data are mechanistically rich and preclinically encouraging. The pancreatitis case is a reminder that expanding indications and expanding use will, statistically, surface rarer adverse events more often. None of this changes day-one prescribing. All of it should change how curious readers interpret the next round of headlines.
If you're tracking this space because you take one of these molecules — or are thinking about it — the work to do is the boring kind. Know your baseline labs. Know your family history of pancreatitis and gallbladder disease. Have a real prescriber, not an online questionnaire. And treat preclinical mechanism papers as a map of where the field is headed, not a prescription pad.
Frequently asked questions
What did researchers find when rats were given semaglutide before 5-FU chemotherapy?
Pretreated rats showed lower hepatic enzymes, less oxidative stress, fewer inflammatory markers, and cleaner histology than controls that received 5-FU alone. The authors propose the protection worked through enhanced PINK1/Parkin-mediated mitophagy and suppression of the ROS/NF-κB/NLRP3 inflammasome axis. When the autophagy inhibitor chloroquine was added, the protective effect was abolished.
What is exendin-4, and what did the new study suggest about its role in arterial plaque?
Exendin-4 is a peptide derived from lizard venom that gave the GLP-1 drug class its first foothold. In both macrophage cell models and ApoE-/- mice on a high-fat diet, exendin-4 reduced foam-cell formation and inflammation by regulating a TREM2–JAK2/STAT3 signaling axis — a pathway that drives the build-up of arterial plaque.
What happened to the patient described in the tirzepatide case report?
A 32-year-old woman started tirzepatide for weight loss and, five weeks later, presented with three weeks of worsening epigastric pain, nausea, vomiting, and constipation; her lipase measured 11,645 U/L and imaging confirmed acute interstitial oedematous pancreatitis. Clinicians stopped tirzepatide, managed her conservatively, and her lipase normalized by discharge. They classified it as a probable case of tirzepatide-induced acute pancreatitis based on the temporal relationship and absence of an alternative culprit.
How is tirzepatide different from semaglutide and exendin-4?
Tirzepatide is a dual GLP-1/GIP agonist, making it chemically distinct from semaglutide and exendin-4, which are GLP-1 receptor agonists. Because it is a newer molecule, the article notes that its post-marketing surveillance picture is still being filled in.
What practical steps does the article suggest for people who take or are considering GLP-1 drugs?
The article advises knowing your baseline labs, knowing your personal and family history of pancreatitis and gallbladder disease, and working with a real prescriber rather than an online questionnaire. It also states that persistent upper abdominal pain on any incretin-class agent is a reason to stop dosing and get a lipase test rather than waiting it out.
Sources
- Exploring novel protective role of semaglutide in 5-fluorouracil-induced hepatotoxicity: Insights into phosphorylated CREB, PINK1/Parkin-mediated mitophagy, and NF-κB/NLRP3 pathways. — The Journal of pharmacology and experimental therapeutics
- Exendin-4 Prevents oxLDL-Induced upregulation of TREM2 and attenuates foam cell formation and inflammation in Macrophages. — Biochemical pharmacology
- A Probable Case of Tirzepatide-Induced Acute Pancreatitis. — Cureus